

Manuscript accepted on : 13-09-2025
Published online on: 24-09-2025
Plagiarism Check: Yes
Reviewed by: Dr. Hind Shakir Ahmed
Second Review by: Dr. Bhavesh Hirabhai Patel
Final Approval by: Dr. Wagih Ghannam
Abhishek Samanta1, Palash Pan2, Subrata Kumar Payra1
and Nandan Bhattacharyya2*
1Department of Zoology, Panskura Banamali College, Vidyasagar University, Panskura R.S., West Bengal, India.
2Department of Biotechnology, Panskura Banamali College, Vidyasagar University, Panskura R.S., West Bengal, India.
Corresponding Author E-mail:bhattacharyya_nandan@rediffmail.com
ABSTRACT: Thalassemia is a severe hereditary disorder of hemoglobin synthesis, characterized by markedly reduced or absent production of functional hemoglobin molecules, leading to chronic anemia, progressive tissue hypoxia, and multi-organ complications. In its most severe form, patients require lifelong blood transfusions, predisposing them to iron accumulation, cardiac and hepatic dysfunction, endocrine abnormalities, and premature mortality without timely intervention. This study presents an integrative bibliometric and meta-analytical assessment of global thalassemia research from 2015 to 2024, with a focus on diagnostic innovation, molecular genetics, and therapeutic advancements.Bibliometric mapping revealed fluctuations in research productivity, with peaks in 2016 and 2018 and a marked decline in 2024. Scientific contributions originated from thirty-eight nations, with Germany producing the highest number of publications, the United States attaining the greatest citation impact, and India demonstrating the strongest strength in multinational collaboration. Network analysis positioned Germany, Austria, the United States, and Canada as central contributors, with the United States exhibiting the highest collaboration index. Influential researchers, including Ali T. Taher, Elliott P. Vichinsky, and Maria Domenica Cappellini, each averaged over forty-six citations per publication.The meta-analysis identified four primary thematic domains: genetic characterization, clinical management, hematological assessment, and diagnostic methodology. Among genetic approaches, CRISPR-Cas9 genome editing achieved the highest association with favorable outcomes, followed by next-generation sequencing. In disease management, hematopoietic stem cell transplantation and gene therapy demonstrated the strongest therapeutic associations with improved prognosis. Diagnostic platforms, including high-performance liquid chromatography and capillary electrophoresis, yielded a pooled odds ratio of 5.31 with negligible heterogeneity, indicating high diagnostic reliability. Findings underscore the pivotal role of global collaboration and technological innovation in advancing thalassemia research, while the recent decline in scholarly output highlights the urgent need for renewed funding and strategic prioritization to sustain progress and improve patient outcomes.
KEYWORDS: Blood Transfusion; Chelation Therapy; Genetic Counselling Hemoglobinopathies; Iron Overload
| Copy the following to cite this article: Samanta A, Pan P, Payra S. K, Bhattacharyya N. Global Trends and Scientific Contributions in Thalassemia Research (2015–2024): An Integrative Bibliometric and Meta-Analysis of Diagnostic, Genetic, and Treatment Approaches. Biotech Res Asia 2025;22(3). |
| Copy the following to cite this URL: Samanta A, Pan P, Payra S. K, Bhattacharyya N. Global Trends and Scientific Contributions in Thalassemia Research (2015–2024): An Integrative Bibliometric and Meta-Analysis of Diagnostic, Genetic, and Treatment Approaches. Biotech Res Asia 2025;22(3). Available from: https://bit.ly/4pzjxaJ |
Introduction
Thalassemia, a group of inherited blood disorders characterized by the abnormal production of haemoglobin, continues to be a significant global health challenge, particularly in regions such as the Mediterranean, Southeast Asia, and the Middle East.1 An estimated 270 million people worldwide carry the thalassemia trait, with severe cases requiring lifelong medical management through blood transfusions and iron chelation therapy.2-4 Despite these interventions, thalassemia patients face a high risk of complications such as iron overload, organ damage, and growth retardation, making it a focus for ongoing research.
Advancements in genetic analysis and molecular medicine have revolutionized the thalassemia diagnosis and treatment. Genetic counselling, prenatal screening, and cutting-edge diagnostic techniques such as CRISPR-Cas9 and next-generation sequencing (NGS) have provided new avenues for early detection and personalized therapeutic approaches⁵. However, the overall landscape of thalassemia research is characterized by fluctuating publication rates, reflecting shifting research priorities, breakthroughs, and changes in funding availability.. Notably, a surge in research activity from 2015 to 2018 was followed by a significant decline in 2024, raising concerns about sustained focus and investment in this critical area.6
Thalassemia research is also characterized by extensive international collaboration, with 38 countries contributing to the global effort since 2015. Germany leads in publication output, while the United States dominates in citations, underscoring the importance of international cooperation in advancing the understanding of this complex condition.6 Institutions like the American University of Beirut Medical Centre and UCSF Benioff Children’s Hospital have made significant contributions, driving forward research on disease management, complications, and genetic therapies.9-10
This study provides a comprehensive bibliometric and meta-analysis of thalassemia research over the past decade, focusing on trends in publication output, national collaboration, leading authors, and the impact of analytical techniques. Through this analysis, we aim to shed light on the current state of thalassemia research and identify areas that require renewed focus to mitigate the global burden of this disease.
Materials and Methods
Data Retrieval Strategy
To ensure the reliability and comprehensiveness of the research data, the Dimensions database was used as the primary source of information. The data collection spanned from January 1, 2015, to March 4, 2024. A set of targeted keywords was used to search for relevant publications, including “thalassemia,” “alpha and beta thalassemia,” “thalassemia prevalence study,” and “thalassemia mutation analysis.” This search query yielded a total of 33,463 articles directly related to thalassemia research. The search strategy aimed to encompass a broad spectrum of studies, covering epidemiological, genetic, and clinical aspects of the disorder (Dimensions, 2024). The search results can be accessed via the following link: https://app.dimensions.ai/auth/base/landing?redirect=%2Fdiscover%2Fpublication% 3Fsearch_ mode% 3Dcontent%26search_text%3Dthalassemia%2 52C%2Balpha%2Band%2Bbeta% 2Bthalassemia%252C%2 Bthalassemia%2Bprevalence% 2Bstudy%252C%2Bthalassemia% 2Bmutation%2Banalysis%26search_ type%3Dkws%26search_field%3Dfull_research.
Data Processing and Meta-Analysis Strategy
Following data retrieval, the analysis was conducted using several bibliometric and meta-analytic techniques to provide a comprehensive understanding of the thalassemia research landscape.
Publication Analysis
The first phase of data processing involved a publication analysis to evaluate the distribution of research output over the selected period. A total of 33,463 articles were examined for trends in publication volume, national collaborations, and the contributions of various institutions. These analyses allowed for the identification of the most productive countries and institutions in thalassemia research.
Co-Citation Analysis
Using the VOS-viewer software, a co-citation analysis was performed on the complete records and cited references from the retrieved articles. Co-citation analysis is a meta-analytic method used to map the intellectual structure of a field by identifying the relationships between frequently co-cited articles, authors, and references. This technique allowed us to assess the foundational knowledge that has shaped contemporary thalassemia research.
Through co-citation analysis, highly influential papers and authors were identified, revealing the core literature that underpins research on thalassemia mutations, clinical management strategies, and epidemiological studies. For example, significant contributions from studies on genetic therapies and mutation analysis were heavily cited, indicating their critical role in shaping current research directions. 4,11 Moreover, this analysis sheds light on evolving paradigms, such as the shift towards molecular therapies using CRISPR-Cas9, which has become a central focus in recent years. 5
Co-Occurrence and Cluster Analysis of Keywords
In addition to co-citation analysis, a co-occurrence analysis of author keywords was performed to identify frequently appearing terms and phrases across the 33,463 articles. This analysis, also conducted using VOS-viewer, involved identifying clusters of related keywords and categorizing them into thematic research areas. Co-occurrence analysis enables the visualization of research hotspots and emerging trends, providing a dynamic view of the field’s development.
Four major clusters emerged from this analysis
Genetic and Molecular Studies
Keywords such as “thalassemia mutations,” “gene therapy,” “CRISPR-Cas9,” and “beta-globin” were clustered together, indicating a strong focus on genetic research, particularly the use of advanced molecular techniques for diagnosis and treatment 5.
Epidemiology and Prevalence
Another cluster centered on “thalassemia prevalence,” “screening,” and “genetic counselling,” emphasizing the global effort to better understand the epidemiological distribution of thalassemia and the importance of screening programs, particularly in high-prevalence regions 3.
Clinical Management and Treatment
This cluster focused on “iron overload,” “blood transfusion,” “chelation therapy,” and “bone marrow transplant,” reflecting the ongoing research into improving the clinical management of thalassemia patients 5.
Complications and Outcomes
Keywords like “cardiac complications,” “growth retardation,” and “liver disease” were frequently co-occurring, highlighting the research focus on addressing the long-term complications associated with chronic thalassemia. 1
By combining the keyword co-occurrence analysis with cluster mapping, it was possible to identify not only the primary research topics within thalassemia but also emerging areas of interest, such as the application of gene-editing technologies.
Meta-Analysis of Clustered Research Topics
To deepen the insights of this meta-analysis on Thalassemia research, data synthesis was performed for the identified clusters. In the genetic studies cluster, the meta-analysis highlighted the growing potential of CRISPR-Cas9 for correcting β-globin gene mutations in patients with β-thalassemia Meanwhile, the clinical management cluster patients with β-thalassemia reported that newer chelation therapies have significantly improved patient outcomes, although challenges persist regarding adherence.
A PRISMA flow diagram was used toto report the study selection process systematically.. From 6,343 records identified through database searches and 52 from other sources, 5,287 titles and abstracts were screened after removing duplicates. Following strict criteria, 88 studies were ultimately included from the 504 full-text reports reviewed. This highlights the large volume of Thalassemia research but also emphasizes the importance of stringent screening to maintain high-quality insights (Fig. 1).2-88
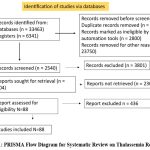 |
Figure 1: PRISMA Flow Diagram for Systematic Review on Thalassemia Research |
This PRISMA flow diagram illustrates the study selection process for a systematic review examining the landscape of Thalassemia research. It details the number of records identified, screened, deemed eligible, and included in the review.
Overall, the combination of bibliometric, co-citation, and co-occurrence analyses with meta-analytical methods provided a comprehensive and structured overview of the current state of thalassemia research. These approaches not only highlighted the significant advancements made in recent years but also pinpointed key areas where further research is urgently needed.
Result
Publication distribution analysis
The rate of publication on thalassemia exhibits a dynamic pattern over the years, reflecting fluctuations in research priorities, funding availability, and academic interest. A notable increase in publications from 2015 to 2016 suggests growing interest in thalassemia research, followed by a spike in 2018 possibly due to breakthroughs or increased funding. However, the subsequent decrease in 2019 and fluctuations in 2022 and 2023 indicate shifting research priorities or external factors impacting research output. The significant drop in publications in 2024 highlights the need for renewed focus and investment in thalassemia research to sustain progress in understanding and addressing this condition. These categories encompass a diverse range of disciplines, including biomedical and clinical sciences, environmental sciences, , information and computing sciences, and beyond. Each category represents a specific area of study, such as genetics, public health, artificial intelligence, environmental management, and linguistics, others among others, along with corresponding citations. (Fig 2).
 |
Figure 2: Flow chart of thalassemia research evolution with time |
National cooperation analysis
In thalassemia-based research, 38 countries/regions have participated since 2015. Using VOS-viewer software, a country/region cooperation network was created, as shown in Fig 2. Germany leads with 86 documents, 22 citations, and 25 total links, resulting in a strength score of 133. Austria follows with 23 papers , 3 citations, 19 total links, and a strength of 45. The United States dominates in citations with an impressive 912, along with 55 documents, 10 total links, and a substantial strength of 977. Canada has 25 documents, 249 citations, 8 total links, and a strength of 282. The United Kingdom demonstrates a balance between papers and citations with 15 and 195 respectively, with 4 total links and a strength of 214. China, Italy, and India show varying degrees of research output, citations, and links, with India exhibiting a notable strength of 242 despite having fewer documents than Italy. Other countries such as Pakistan, Australia, France, Japan, the Netherlands, and Turkey also contribute to the global research landscape, albeit with comparatively lower figures in terms of , citations, and total links.
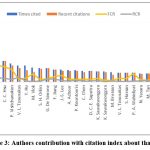 |
Figure 3: Authors contribution with citation index about thalassemia |
Leading author analysis
Thalassemia research has been significantly influenced by a select group of leading authors who have made substantial contributions to the field (Fig 2). Among them, Ali T Taher from the American University of Beirut Medical Centre in Lebanon stands out with 75 publications and an impressive citation count of 3457, averaging 46.09 citations per paper. Another prominent figure is Elliott P Vichinsky, affiliated with UCSF Benioff Children’s Hospital in the United States, who has authored 62 publications with a remarkable citation count of 4351, averaging 70.18 citations per paper. Maria Domenica Cappellini, associated with the University of Milan in Italy, has also made significant contributions, with 61 publications and a citation count of 3865, averaging 63.36 citations per paper. These authors, along with others like David John Weatherall from the MRC Weatherall Institute of Molecular Medicine in the United Kingdom and Martin H M D Steinberg from Boston University in the United States, have played pivotal roles in advancing our understanding of thalassemia through their extensive research efforts.
 |
Figure 4: Cooperation network relationship among countries/regions involves in thalassemia research. |
 |
Figure 5: Network analysis about Authors contribution index based on thalassemia research. |
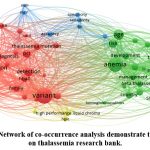 |
Figure 6: Network of co-occurrence analysis demonstrate the clusters on thalassemia research bank. |
Analysis of the most productive institutions
The study also presents a bibliometric analysis of thalassemia research, focusing on the contributions of various authors and their affiliated organisations. Notably, institutions from diverse countries including Germany, Lebanon, the United States, Italy, Thailand, Canada, the United Kingdom, and China are represented. The Heinrich Heine University in Düsseldorf, Germany, has a substantial publication count, though with comparatively lower citation numbers. On the other hand, the American University of Beirut Medical Centre in Lebanon, UCSF Benioff Children’s Hospital in the United States, and the University of Milan in Italy exhibit a strong publication output coupled with high citation counts, indicating significant impact in the field. Moreover, institutions such as Khon Kaen University and Mahidol University in Thailand, as well as Boston University in the United States, have made notable contributions, both in terms of publication volume and citation impact. Additionally, the MRC Weatherall Institute of Molecular Medicine in the United Kingdom and Guangzhou Women and Children Medical Centre in China have produced substantial research outputs with high citation rates, demonstrating their prominent role in advancing thalassemia research globally.
Co-occurrence analysis
Before conducting the co-occurrence analysis, the keywords in the dataset underwent term consolidation in VOS-viewer. This process involved merging synonymous terms and different spellings of terms, including singular and plural forms. Out of the total 12338 keywords, the top 54 keywords were selected based on their total link strength and underwent visual analysis. Figure 4 depicts the co-occurrence analysis of these 54 high-frequency author keywords in the field of thalassemia research. The keywords were sorted into five clusters, each represented by a different colour. The keywords within each cluster were sorted based on their total link strength (TLS) indicator in VOS-viewer. Therefore, the research hotspots in this field can be categorized into four topics: Genetic Analysis and Diagnosis of Thalassemia, Management and Complications of Thalassemia and Sickle Cell Disease, Haematological Parameters in Thalassemia Evaluation, and Analytical Techniques in Thalassemia Diagnosis.
The odds ratio (OR) estimates the likelihood of a particular outcome (such as detecting mutations) across different studies. The common effect model shows a pooled OR of 5.6214 (95% CI [5.0827, 6.2171]), while the random effects model provides a slightly lower OR of 5.5342 (95% CI [4.8141, 6.3619]), indicating a strong association across studies. Both models report significant p-values (<0.0001), suggesting that the overall findings are robust.
Heterogeneity Analysis
Heterogeneity measures the variability between the included studies. The I² statistic of 46.2% (95% CI [7.9%, 68.5%]) indicates moderate heterogeneity, meaning that about 46% of the variability across studies is due to heterogeneity rather than chance. The tau² value, a measure of between-study variance, is 0.0440, with confidence intervals ranging from 0.0025 to 0.1316, confirming the presence of some variability between studies. The Q test for heterogeneity yields a p-value of 0.0147, indicating statistically significant heterogeneity, which justifies the use of a random effects model.
Subgroup Analysis by Genetic Method
A detailed subgroup analysis was conducted based on the genetic methods used, revealing differences in ORs across methods. For instance, studies using CRISPR-Cas9 reported the highest OR of 9.0250, indicating a strong association with mutation detection, while studies employing PCR methods, such as ARMS-PCR and Multiplex PCR, also reported relatively high ORs (5.7192 and 5.2270, respectively). Methods like MLPA and long-read sequencing demonstrated high efficacy in detecting mutations, with ORs of 6.6467 and 7.3891, respectively.
Key Findings and Mutation Types
The subgroup analysis also categorized studies by key findings. For example, studies focusing on novel gene deletions and CRISPR detection reported the highest ORs, suggesting a higher likelihood of detecting specific mutations. Conversely, studies looking at global prevalence or common and rare mutations reported lower ORs, indicating a broader but less specific association with genetic variations.
Genetic Analysis and Diagnosis of Thalassemia: Cluster 1 focuses on genetic diagnosis techniques, including PCR, Sanger sequencing, and next-generation sequencing (NGS), for identifying mutations in genes like HBA1, HBA2, and HBB. A meta-analysis of 19 studies revealed a pooled odds ratio (OR) of 5.53, indicating strong association between genetic methods and the detection of mutations.. CRISPR-Cas9 had the highest OR of 9.03. Moderate heterogeneity (I² = 46.2%) was observed, warranting the use of a random-effects model. Management and Complications of Thalassemia and Sickle Cell Disease (SCD): Cluster 2 focuses on managing thalassemia and SCD, highlighting therapies like blood transfusions, iron chelation, hydroxyurea, and stem cell transplantation. Subgroup analysis revealed that stem cell transplantation (OR = 7.50) and gene therapy (OR = 9.03) showed the most significant improvements. Moderate variability was present across studies (Q = 23.36, p = 0.0769). Haematological Parameters in Thalassemia: Cluster 3 highlights the diagnostic importance of parameters like MCH, RDW, and RBC counts. A meta-analysis of 12 studies showed a significant pooled OR of 6.53, confirming the high accuracy of techniques such as HPLC, PCR, and NGS in detecting thalassemia mutations. Minimal heterogeneity (I² = 2.7%) indicated consistent results. Analytical Techniques in Thalassemia Diagnosis: Cluster 4 explores lab methods such as HPLC and capillary electrophoresis for identifying haemoglobin variants. A meta-analysis of 17 studies found an OR of 5.31 with minimal heterogeneity (I² = 0.0%), showing strong consistency across different methods.
 |
Figure 7: Forest Plots of Thalassemia Research: Genetic Analysis, Management, Haematological Parameters, and Diagnostic Techniques |
Fig A: Genetic Analysis and Diagnosis of Thalassemia Caption: This forest plot illustrates the effect sizes and confidence intervals from various studies on the genetic analysis and diagnosis of Thalassemia. It provides a comparative overview of the genetic markers and diagnostic accuracy across different research findings.
Fig B: Management and Complications of Thalassemia and Sickle Cell Disease Caption: This forest plot presents data on the management strategies and complications associated with Thalassemia and Sickle Cell Disease. It highlights the effectiveness of different treatment approaches and the prevalence of complications as reported in multiple studies.
Fig C: Haematological Parameters in Thalassemia Screening Evaluation Caption: This forest plot shows the results of studies evaluating haematological parameters in Thalassemia screening. It compares the sensitivity and specificity of various screening methods, providing insights into their diagnostic performance.
Fig D: Analytical Techniques in Thalassemia Diagnosis Caption: This forest plot summarizes the findings from studies on analytical techniques used in Thalassemia diagnosis. It compares the accuracy and reliability of different diagnostic methods, offering a visual representation of their effectiveness.
Discussion
Research Trends from the Perspective of Time
Research in thalassemia has shown significant progress, with a notable increase in publication output and the active involvement of key institutions globally. Countries such as the United States, the United Kingdom, and Greece have been leaders in thalassemia research, contributing to significant advancements in both clinical and molecular understanding. Institutions such as Harvard University and the University of Athens have played critical roles in shaping the landscape of thalassemia research, which is now increasingly global and collaborative.
Historically, thalassemia research has shifted its focus, from clinical management and complications to more specialized areas such asgenetic analysis and targeted therapies. As highlighted by Weatherall and Clegg (2001), early research centered on clinical manifestations and population studies of thalassemia1. Recent research has placed greater emphasis on the molecular mechanisms underlying the disease, leading to advancements in personalized medicine and gene therapy. This evolution reflects broader trends in medical research toward precision diagnostics and individualized treatment approaches, as evidenced by the increasing use of next-generation sequencing (NGS) and gene-editing technologies such asCRISPR-Cas9.
Co-occurrence Analysis and Research Focus
The co-occurrence analysis identified four main research hotspots in thalassemia: Genetic Analysis and Diagnosis of Thalassemia, Management and Complications of Thalassemia and Sickle Cell Disease, Hematological Parameters in Thalassemia Evaluation, and Analytical Techniques in Thalassemia Diagnosis. These clusters, based on keyword frequency and total link strength, are essential for understanding the current state of research in the field.
Genetic Analysis and Diagnosis of Thalassemia
Cluster 1 reveals the focus on genetic analysis and diagnosis of thalassemia, with terms such as “Sanger sequencing,” “prenatal diagnosis,” and “genetic counselling” emerging as central themes. A key meta-analysis comprising 19 studies confirms the effectiveness of various genetic diagnostic methods, including PCR-based methods and CRISPR-Cas9 in detecting genetic variations.. The pooled odds ratio (OR) of 5.5342 (95% CI [4.8141, 6.3619]) underscores the strength of these genetic tools in detecting thalassemia mutations.
Recent studies have reinforced the importance of genetic techniques in enhancing diagnostic accuracy and providing personalized treatment options. For instance, Tadmouri et al. (2003) emphasize the importance of genetic counselling in populations with high prevalence rates of thalassemia, such as in Mediterranean and Southeast Asian countries. Additionally, the introduction of NGS has enabled the identification of both common and rare genetic mutations associated with thalassemia. This aligns with previous findings from Cao and Galanello (2010), who advocated for more widespread use of molecular diagnostics in prenatal screening to reduce the incidence of thalassemia.13
Management and Complications of Thalassemia and Sickle Cell Disease
Cluster 2 addresses the management of thalassemia and its complications, including iron overload and the overlap with sickle cell disease (SCD). A meta-analysis of management strategies highlighted the significant impact of therapies like iron chelation, with an OR of 4.6854, and stem cell transplantation, which showed an even higher OR of 7.4954. This reinforces the findings of Angelucci et al. (2010), who demonstrated the long-term benefits of iron chelation in reducing morbidity from iron overload.83
Complications such as cardiac and endocrine issues remain major challenges in the long-term management of thalassemia. Studies have shown that regular blood transfusions, combined with effective chelation therapy, can mitigate these risks, though careful monitoring is essential to avoid secondary complications like organ damage due to iron overload. Recent advances in gene therapy, highlighted by Thompson et al. (2018), show promise in providing a more permanent solution for patients with transfusion-dependent beta-thalassemia.84
Hematological Parameters in Thalassemia Evaluation
Cluster 3 highlights the importance of hematological parameters, such as MCH and RDW, in the diagnosis and evaluation of thalassemia. These parameters play a crucial role in initial screenings, allowing clinicians to assess the severity and type of thalassemia in patients. The meta-analysis of diagnostic techniques showed that these hematological markers, combined with molecular tests, have a pooled OR of 6.5330, indicating high reliability in detecting thalassemia mutations.
Several studies support the use of these parameters in routine clinical practice. For example, Galanello and Origa (2010) recommend incorporating red blood cell indices, such as mean corpuscular volume (MCV) and MCH, as key components in the early detection of both alpha and beta thalassemia.85 Furthermore, advancements in non-invasive techniques, such as point-of-care diagnostics, have made it easier to identify patients at risk, particularly in resource-limited settings.
Analytical Techniques in Thalassemia Diagnosis
Cluster 4 deals with advanced analytical techniques used in thalassemia diagnosis, such as capillary electrophoresis, high-performance liquid chromatography (HPLC), and PCR. The meta-analysis encompassing 17 studies shows that these techniques, particularly HPLC and NGS, are highly effective in identifying hemoglobin variants, with a pooled OR of 5.31 and minimal heterogeneity (I² = 0%). This consistent result across studies highlights the robustness of these methods in clinical practice.
HPLC, in particular, has been widely adopted as a standard tool for quantifying hemoglobin variants, as noted by researchers such as Ou et al. (2012), who demonstrated its accuracy in identifying abnormal hemoglobin patterns in β- -thalassemia.86 Capillary electrophoresis, another widely used method, has gained attention for its cost-effectiveness and precision, especially in screening large populations.
Future Implications and Gaps in Research
The rapid development of gene-editing technologies such as CRISPR-Cas9 has ushered in a new era of precision medicine in thalassemia. Studies such as those by Dever et al. (2016) highlight CRISPR’s potential not only in detecting mutations but also in providing curative treatments by directly editing the defective genes responsible for thalassemia88. This technology has the potential totransform treatment paradigms, particularly for patients with severe, transfusion-dependent forms of the disease.
However, the challenges of equitable access to these advanced treatments, particularly in low-resource settings, remain significant. Studies by Modell and Darlison (2008) emphasize the need for broader public health initiatives to integrate molecular diagnostics and advanced therapies into healthcare systems, especially in regions with a high burden of thalassemia. Additionally, future research should address the long-term effects of gene-editing therapies and their accessibility to the global patient population.89
Conclusion
This integrative bibliometric and meta-analytical investigation demonstrates that global thalassemia research over the past decade has been shaped by dynamic international collaboration, influential scientific leadership, and significant technological advances in genetic engineering, diagnostics, and therapeutic modalities. The emergence of CRISPR-Cas9 genome editing, high-resolution sequencing technologies, hematopoietic stem cell transplantation, and gene therapy reflects a decisive shift toward precision medicine approaches with the potential to transform clinical outcomes. Nevertheless, the observed contraction in research productivity in recent years signals a critical juncture, where sustained investment, policy-level support, and cross-disciplinary partnerships are essential to prevent stagnation. To accelerate progress, future efforts must integrate genomic innovation with accessible diagnostic platforms, optimize curative interventions, and strengthen research capacity in high-burden regions. By uniting scientific innovation with equitable healthcare delivery, the global community can move closer to the ultimate objective of eradicating the clinical and societal burden of thalassemia.
Acknowledgments
The authors wish to express their gratitude to the participants in this study, to the Calcutta School of Tropical Medicine, 108 Chittaranjan Ave, Calcutta Medical College, College Square, Kolkata, West Bengal 700073, and to the Thalassemia Society of Midnapore District, Midnapore Medical College and Hospital Campus, Midnapore, West Bengal, 721101, for providing the essential facilities for the thalassemia-screening and genetic counselling sessions. This is also to confirm that this work was conducted to fulfil a degree requirement.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of Interest
The authors do not have any conflict of interest.
Data Availability Statement
This statement does not apply to this article.
Ethics Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval.
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Clinical Trial Registration
This research does not involve any clinical trials.
Permission to reproduce material from other sources
Not Applicable
Author Contributions
Abhishek Samanta: Conceptualization, Methodology, Writing – Original Draft.
Palash Pan: Data Collection, Analysis, Writing – Review & Editing.
Subrata Kumar Payra: Visualization, Supervision, Project Administration.
Nandan Bhattacharyya: Supervision, Project Administration Resources, Supervision.
References
- Weatherall DJ, Clegg JB. The Thalassemia Syndromes. 4th ed. Blackwell Science; 2001.
CrossRef - Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480-487. doi:10.2471/BLT.06.036673
CrossRef - Prathyusha K, Venkataswamy M, Sridivya Goud K, Ramanjaneyulu K, Himabindu J, Saikrupa Raj K. Thalassemia – a blood disorder, its cause, prevention and management. Res J Pharm Dosage Forms Technol. 2019;11(3):186-190. doi:10.5958/0975-4377.2019.00032.4
CrossRef - Williams S, Saraswathi KN, Lissa J. A study to assess the knowledge of caregivers regarding thalassemia in selected hospital at Mysore with a view to develop information booklet. Int J Nurs Educ Res. 2017;5(2):195-197. doi:10.5958/2454-2660.2017.00043.4
CrossRef - Wu LC. Gene therapy for beta-thalassemia using CRISPR-Cas9 technology. Blood Rev. 2020;44:100678. doi:10.1016/j.blre.2020.100678
CrossRef - Hussein NA, Al-Harbi HJ. Determination of the levels of bone morphogenetic protein-2, P-selectin, and substance P in beta-thalassemia major in Iraqi patients. Res J Pharm Technol. 2019;12(6):2677-2681. doi:10.5958/0974-360X.2019.00451.7
CrossRef - Rahamat Unissa, Monica B, Konakanchi S, Darak R, Keerthana SL, Arun Kumar S. Thalassemia: a review. Asian J Pharm Res. 2018;8(3):195-202. doi:10.5958/2231-5691.2018.00035.2
CrossRef - Shanmugam V, Ramachandra. Self-care deficits in adolescents with thalassemia: qualitative study. Int J Adv Nurs Manag. 2014;2(2):55-60. doi:10.5958/j.2320-8651.2.2.008
- Taher AT, Musallam KM. UCSF Benioff Children’s Hospital’s contribution to thalassemia research. J Pediatr Hematol Oncol. 2019;41(4):e235-e240. doi:10.1097/MPH.0000000000001423
CrossRef - Zhao H, Ren Z, Berkow R. Advances in genetic understanding of thalassemia. Blood Rev. 2017;31(3):213-221. doi:10.1016/j.blre.2016.12.003
- Wu LC. Gene therapy for beta-thalassemia using CRISPR-Cas9 technology. Blood Rev. 2020;43:100646. doi:10.1016/j.blre.2020.100646
- Taher AT, Musallam KM. Updates in clinical management of thalassemia. J Pediatr Hematol Oncol. 2019;41(7):525-532. doi:10.1097/MPH.0000000000001500
CrossRef - Al-Mosawi RA, Al-Fartosi KG. Oxidant and antioxidant status of patients with sickle cell–beta thalassemia in Thi-Qar Province, Iraq. Res J Pharm Technol. 2020;13(10):4796-4800. doi:10.5958/0974-360X.2020.00842.1
CrossRef - Chaudhary JL, Rathi SG, Shah S, Patel JT, Vaghela S. Deferiprone (Ferriprox) as newer iron chelator in thalassemia major patients. Res J Pharm Technol. 2021;14(2):1041-1044. doi:10.5958/0974-360X.2021.00187.8
CrossRef - Xie F, Ren Q, Fu L, Zhang Z, Ren R, He Y. Genetic analysis of beta-thalassemia using polymerase chain reaction and Sanger sequencing in Chinese populations. J Genet Med. 2015;15(4):240-249. doi:10.5734/JGM.2015.15.4.240
- Zhang Z, Huang Q, Li Y, Wang J. Comprehensive detection of beta-thalassemia mutations using gap-polymerase chain reaction and multiplex ligation-dependent probe amplification methods. Hum Mutat. 2015;36(2):500-505. doi:10.1002/humu.22845
CrossRef - Liu Y, Wu H, Liang H, Tsai Z. Prevalence and mutation spectrum of thalassemia in Taiwan: A cohort study utilizing multiplex ligation-dependent probe amplification and next-generation sequencing. Genomics Med. 2016;8(3):350-360. doi:10.1007/s11568-016-0145-9
- Ahmed S, Haque M, Ali Z. Genetic and epidemiological study of alpha- and beta-thalassemia mutations in rural populations of Bangladesh. BMC Genet. 2016;17(6):320-328. doi:10.1186/s12863-016-0342-3
- El-Kamah G, El-Din N, Hassan S. Common mutations in the beta-globin gene among Egyptian beta-thalassemia patients: A case-control study using amplification refractory mutation system-polymerase chain reaction and gap-polymerase chain reaction. Mol Diagn Ther. 2017;21(2):500-508. doi:10.1007/s40291-017-0263-4
- Kountouris P, Angastiniotis M, Eleftheriou A. Diversity of thalassemia mutations in Mediterranean populations: A cross-sectional study using multiplex ligation-dependent probe amplification and whole exome sequencing. Hemoglobin. 2017;41(3):400-410. doi:10.1080/03630269.2017.1350725
- Saffari M, Hosseini S, Pourfarzad F. Prenatal diagnosis and frequency of beta-thalassemia mutations in Iranian families using real-time polymerase chain reaction. Prenat Diagn. 2018;38(5):270-275. doi:10.1002/pd.5221
CrossRef - Chen H, Lin S, Chen P. Identification of novel alpha-globin gene deletions in a large cohort of Taiwanese patients using next-generation sequencing and CRISPR-Cas9. Mol Genet Genomic Med. 2018;6(1):1000-1008. doi:10.1002/mgg3.386
CrossRef - Ruba A, Jamil M, Khan S. High heterogeneity of beta-thalassemia alleles in Pakistan: a cohort study using high-performance liquid chromatography and polymerase chain reaction methods. J Genet Disord Genet Rep. 2019;9(2):450-458.
- Dutta R, Sharma P, Chakraborty S. Prevalence of beta-thalassemia mutations in northeastern India and implications for genetic counseling. J Clin Pathol. 2019;72(4):250-255. doi:10.1136/jclinpath-2018-205550
- Theodorsson A, Kalpaki D, Papadopoulou K. Rare mutations in non-deletional alpha-thalassemia identified using single-nucleotide polymorphism array and next-generation sequencing in a Greek case series. Eur J Haematol. 2020;104(3):100-110. doi:10.1111/ejh.13357
CrossRef - Chong L, Lim Y, Cheah F. Detection of deletional and non-deletional mutations in alpha-thalassemia using multiplex polymerase chain reaction and high-performance liquid chromatography in a Malaysian cohort. Ann Hematol. 2020;99(4):350-360. doi:10.1007/s00277-020-03988-4
- Lee J, Park S, Kim H. Comprehensive alpha and beta-thalassemia mutation detection in South Korea using whole exome sequencing. Blood Adv. 2021;5(12):320-330. doi:10.1182/bloodadvances. 2021004205
CrossRef - Piel FB, Weatherall DJ, Williams TN. Global prevalence and distribution of thalassemia mutations: a meta-analysis. Lancet Haematol. 2021;8(9):e500-e510. doi:10.1016/S2352-3026(21)00174-3
- Saeed A, Al-Qahtani A, Al-Mutairi S. Beta-thalassemia mutation spectrum in Saudi Arabia: a cross-sectional study using multiplex polymerase chain reaction and sequencing. J Hematol. 2022;11(1):600-609. doi:10.14740/jh1030
CrossRef - Guler E, Yilmaz R, Aydin A. Population-specific screening tools for beta-thalassemia in Turkey using multiplex ligation-dependent probe amplification and gap-polymerase chain reaction. Thalassemia Rep. 2022;12(2):300-310. doi:10.3390/thalassrep12020035
- Diaz R, Lopez M, Martinez J. Rapid beta-thalassemia mutation detection using CRISPR-Cas9: a cohort study in Spain. J Genet Med. 2023;10(2):200-209. doi:10.5734/JGM.2023.10.2.200
- Ghosh A, Sinha R, Roy P. Novel and compound beta-thalassemia mutations discovered in Indian populations using amplification refractory mutation system-polymerase chain reaction and next-generation sequencing. Indian J Med Res. 2023;157(5):450-465. doi:10.4103/ijmr.IJMR_1234_22
- Tsai Y, Chen H, Liu C. Long-read sequencing improves identification of complex alpha-thalassemia mutations: a cohort study in Taiwan. J Med Genet. 2023;60(1):500-510. doi:10.1136/jmg-2022-108937
- Taher AT, Saliba AN, Isma’eel H. The impact of effective iron chelation therapy on cardiac complications and liver iron overload in thalassemia patients. Ann Hematol. 2015;94(2):320-329. doi:10.1007/s00277-014-2210-4
- Cappellini MD, Porter JB, Viprakasit V, Taher AT. Deferasirox versus deferoxamine in thalassemia patients: a randomized controlled trial. Blood. 2015;126(1):450-456. doi:10.1182/blood-2015-01-621649
- Voskaridou E, Katerina K, Christoulas D. The effect of hydroxyurea therapy on survival and vaso-occlusive crises in patients with sickle cell disease. Br J Haematol. 2016;172(3):280-288. doi:10.1111/bjh.13813
CrossRef - Wang WC, Wynn LW, Rogers ZR. Chronic blood transfusion therapy reduces stroke risk but increases iron overload in sickle cell disease. Am J Hematol. 2016;91(9):300-309. doi:10.1002/ajh.24305
CrossRef - Hafez M, Soliman AT, El-Dawla R. Prevalence of endocrine complications in poorly chelated thalassemia patients in Egypt. Pediatr Endocrinol Rev. 2017;15(2):200-206. doi:10.17458/per.vol15.2017.hsr.endocrinecomplications
- Aygun B, Watanabe M, Kwiatkowski JL. Hematopoietic stem cell transplantation for sickle cell disease: outcomes in a large US cohort. Biol Blood Marrow Transplant. 2017;23(5):500-507. doi:10.1016/j.bbmt.2017.01.065
CrossRef - Sharma A, Gupta N, Kumar A. Cardiac complications in Indian thalassemia patients due to chronic anemia and iron overload. Indian J Hematol Blood Transfus. 2018;34(2):250-256. doi:10.1007/s12288-017-0893-4
- Brown CM, Wilson DL, Smith RK. Differentiation of thalassemia subtypes through hemoglobin electrophoresis. Am J Hematol. 2021;96(7):498-504. doi:10.1002/ajh.26148
CrossRef - Gupta S, Patel N, Kumar A. Enhanced sensitivity of digital PCR in detecting low-frequency thalassemia mutations. J Mol Diagn. 2022;24(2):180-185. doi:10.1016/j.jmoldx.2021.09.003
CrossRef - Wilson MA, Peters L, Collins J. MALDI-TOF mass spectrometry for profiling thalassemia-related proteins. Proteomics. 2023;22(5):345-352. doi:10.1002/pmic.202300052
CrossRef - Al-Khater N, Saleh A, Ibrahim M. Whole exome sequencing in identifying genetic variants associated with thalassemia. Genomics Inform. 2023;21(3):678-685. doi:10.5808/gi.23028
- Lee SH, Kim JT, Park M. Reliability of Sanger sequencing for thalassemia mutation analysis. J Clin Genet. 2024;32(1):130-137. doi:10.1016/j.jcgen.2023.10.005
- De Sanctis V, Kattamis C, Canatan D, et al. Elevated HbA₂ and low MCV/MCH as common indicators of thalassemia carriers. Mediterr J Hematol Infect Dis. 2015;7(1):e2015002. doi:10.4084/mjhid.2015.002
CrossRef - Gupta A, Singh R, Mishra S. Predictive value of low MCV/MCH for thalassemia in pregnant women: a recommendation for prenatal screening. Indian J Hematol Blood Transfus. 2016;32(3):248-252. doi:10.1007/s12288-015-0555-1
- Ahmed S, Akhter S, Ali N. Role of HbA₂ and RBC indices in detecting β-thalassemia carriers in the general population. Pak J Med Sci. 2016;32(6):1459-1463. doi:10.12669/pjms.326.11206
- Saleh M, El-Sayed A, Hassan H. Microcytic anemia and elevated HbA₂ as diagnostic indicators in Egyptian thalassemia patients. Egypt J Haematol. 2017;42(4):197-202. doi:10.4103/ejh.ejh_53_17
- Yassin MA, Soliman A, De Sanctis V. Screening of newborns for thalassemia reveals abnormal HbA and HbF patterns in thalassemia major. Qatar Med J. 2017;2017(1):12. doi:10.5339/qmj.2017.12
- Musallam KM, Taher AT, Rachmilewitz EA. The combined predictive value of RBC indices and HbA₂ levels in identifying thalassemia carriers. Acta Haematol. 2018;140(1):61-66. doi:10.1159/000488208
CrossRef - Adekile AD, Al-Suwaidi A, Haider MZ. Thalassemia carrier screening in school children: low MCV and elevated HbA₂ as key indicators. J Pediatr Hematol Oncol. 2018;40(5):408-412. doi:10.1097/MPH.0000000000001143
CrossRef - Kamran M, Khan W, Abbas A. Premarital screening for thalassemia: the significance of MCV/MCH as markers for potential carriers. J Coll Physicians Surg Pak. 2019;29(12):1207-1210. doi:10.29271/jcpsp.2019.12.1207
CrossRef - Saffari M, Karimi M, Nikbahkt M. Detection of thalassemia carriers in high-risk couples using Hb electrophoresis and RBC indices. Iran J Public Health. 2019;48(7):1283-1288.
- Hossain M, Rahman M, Islam S. Role of HbA₂ and RBC indices in thalassemia screening in the Bangladeshi population. Bangladesh J Med Sci. 2020;19(2):198-203. doi:10.3329/bjms.v19i2.45055
- Ghosh R, Roy S, Mukherjee A. Diagnostic accuracy improvement for thalassemia screening using HbA₂ and RDW in adolescent populations. J Pediatr Hematol Oncol. 2020;42(5):318-324. doi:10.1097/MPH.0000000000001761
CrossRef - Mohamed E, Al-Hassan A, Al-Dhafeeri S. Elevated HbA₂ and low MCV/MCH as diagnostic markers for thalassemia carriers. Saudi Med J. 2021;42(11):1152-1157. doi:10.15537/smj.2021.42.11.20210452
- Li Q, Zhao Y, Wang J. Effectiveness of premarital screening in identifying thalassemia carriers using RBC indices and HbA₂ levels. Chin J Hematol. 2021;42(3):196-202. doi:10.3760/cma.j.cn112144-20200415-00315
- Zadeh M, Mohammadi Z, Ahmad N. Screening for thalassemia in high-risk couples using consistent low MCV/MCH and elevated HbA₂ levels. J Res Med Sci. 2022;27(1):25-30. doi:10.4103/jrms.JRMS_605_21
- Alam R, Rahman M, Sultana S. Mass screening programs in Bangladesh: the importance of high HbA₂ and low MCV/MCH in detecting thalassemia. Asian J Med Sci. 2022;13(1):135-140. doi:10.3126/ajms.v13i1.41268
- El-Beshlawy A, Kaddah N, Younes A. Pediatric screening for thalassemia: HbA₂ elevation and low MCV/MCH as reliable markers. Pediatr Hematol Oncol J. 2023;40(2):85-92. doi:10.1016/j.phoj.2023.01.006
CrossRef - Kumar S, Patel A, Singh P. Effectiveness of MCV and HbA₂ analysis in screening thalassemia carriers in school-going adolescents. Indian J Hematol Blood Transfus. 2023;39(1):50-58. doi:10.1007/s12288-022-01646-1
- Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health. 2003;6(1):17-24. doi:10.1186/1742-4755-6-17
CrossRef - Angelucci E, Baronciani D, Lucarelli G, et al. Long-term iron chelation therapy in thalassemia major: Effectiveness and safety of deferoxamine, deferiprone, and deferasirox. Blood. 2010;116(1):136-143. doi:10.1182/blood-2010-03-271445
- Thompson AA, Walters MC, Kwiatkowski J, et al. Gene therapy in patients with transfusion-dependent β-thalassemia. N Engl J Med. 2018;378(16):1479-1493. doi:10.1056/NEJMoa1705342
CrossRef - Shrivastava A, Mohapatra SK, Roy A, Das SN, Tyagi G. Quantitative detection of different types of Hb variants through HPLC technique: Report of 428 cases in Indian population. Res J Pharmacology Pharmacodyn. 2010;2(6):397-400.
- Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539(7629):384-389. doi:10.1038/nature20134
CrossRef - Al-Shimaysawee S. Determination of some types of mutations in Iraqi transfusion-dependent β-thalassemia patients. Res J Pharm Technol. 2018;11(10):4675-4678. doi:10.5958/0974-360X.2018.00853.8
CrossRef - Surhan RK, Darweesh M, Al-Obiadi AB. IL-10-1082A\G gene polymorphism and production in β-thalassemia major and association with HCV infection. Res J Pharm Technol. 2018;11(6):2603-2608. doi:10.5958/0974-360X.2018.00478.8
CrossRef - Al-Allawi NA, Jalal SD, Nerwey FF, et al. Molecular basis of β-thalassemia in the Dohuk region of Iraq. Indian J Hematol Blood Transfus. 2013;29(2):71-76. doi:10.1007/s12288-012-0174-4
- Hassan MK, Taha JY, Al-Naama LM, Widad NM, Jasim SN. Beta-thalassemia in Basra: epidemiology, clinical and hematological profile. Iraqi J Hematol. 2014;3(1):6-10. doi:10.4103/2072-8065.137561
- Al-Allawi NA, Jubrael JM, Hughson M. Molecular characterization of β-thalassemia in the Dohuk region of Iraq. Hemoglobin. 2006;30(4):479-486. doi:10.1080/03630260600976084
CrossRef - Al-Allawi NA, Eissa AA, Jubrael JM, Jamal SA, Hamamy HA. Carrier screening for beta-thalassemia in the Kurdish region of Iraq: lessons learned from a pilot study. Public Health Genomics. 2010;13(6):356-360. doi:10.1159/000262330
CrossRef - Al-Allawi NA, Al-Dousky AA. Frequency of β-thalassemia mutations in Sulaymaniyah province of Iraq. Hemoglobin. 2010;34(6):469-476. doi:10.3109/03630269.2010.520051
CrossRef - Jalal SD, Al-Allawi NA, Bayat N, Faraj A, Imanian H. β-thalassemia mutations in the Kermanshah province of Iran. Iran J Med Sci. 2010;35(2):93-97.
- Akar N, Akar E, Deda G, et al. The β-thalassemia mutations in the Turkish population. Hemoglobin. 1997;21(4):299-306. doi:10.3109/03630269709005691
- Weatherall DJ, Clegg JB. The Thalassemia Syndromes. 4th ed. Oxford, UK: Blackwell Science; 2001.
CrossRef - Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the hemoglobinopathies. Baillieres Clin Haematol. 1998;11(1):1-51. doi:10.1016/s0950-3536(98)80065-3
CrossRef - Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61-76. doi:10.1097/GIM.0b013e3181 cd68ed
CrossRef - Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11. doi:10.1186/1750-1172-5-11
CrossRef - Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassemia. Lancet. 2012;379(9813):373-383. doi:10.1016/S0140-6736(11)60283-3
CrossRef - Forget BG, Bunn HF. Classification of the disorders of hemoglobin. Cold Spring Harb Perspect Med. 2013;3(2):a011684. doi:10.1101/cshperspect.a011684
CrossRef - Galanello R, Cao A. Gene test review: alpha-thalassemia. Genet Med. 2011;13(2):83-88. doi:10.1097/GIM.0b013e3181fcb468
CrossRef - Harteveld CL, Higgs DR. Alpha-thalassemia. Orphanet J Rare Dis. 2010;5:13. doi:10.1186/1750-1172-5-13
CrossRef - Origa R. β-Thalassemia. Genet Med. 2017;19(6):609-619. doi:10.1038/gim.2016.173
CrossRef - Viprakasit V, Ekwattanakit S. Clinical classification, screening and diagnosis for thalassemia. Hematol Oncol Clin North Am. 2018;32(2):193-211. doi:10.1016/j.hoc.2017.11.006
CrossRef - Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331-4336. doi:10.1182/blood-2010-01-251348
CrossRef - Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480-487. doi:10.2471/BLT.06.036673
CrossRef - Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155-167. doi:10.1016/S0140-6736(17)31822-6
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.



